

















 11 / 52
11 / 52

11
el desarrollo de los medicamentos
genéricos y no se decidió
arbitrariamente, sino tomando como
base el hecho de que esta diferencia
en los parámetros farmacocinéticos
evaluados se admite como el
umbral que marca la ausencia de
consecuencias en la respuesta
farmacológica y, por ello, no posee
relevancia clínica para la inmensa
mayoría de los fármacos.
El test de bioequivalencia, en contra
de lo que muchos consideran,
incorpora requerimientos muy
estrictos para dar la conformidad.
Si los valores medios del ABC
del producto de referencia y del
‘potencial genérico’ difieren en
una pequeña proporción (» 10%)
es muy improbable que ambos
productos puedan ser considerados
bioequivalentes.
Los voluntarios incluidos en el ensayo
de bioequivalencia reciben las 2
formulaciones (referencia y potencial
genérico), administradas a la misma
dosis, evaluándose los valores medios
de los parámetros farmacocinéticos
(ABC y Cmax) y sus desviaciones
estándar (DE) en el ‘potencial
genérico’ para poder establecer
sus correspondientes IC90. Estos
límites deben estar comprendidos
dentro del ± 20 % del valor medio
de cada uno de los parámetros
farmacocinéticos obtenidos en el
medicamento de referencia. En
consecuencia, el criterio utilizado
para establecer la bioequivalencia,
no es una comparación puntual de
valores medios de las formulaciones
test y de referencia, como
frecuentemente es interpretado,
ya que eso supondría aceptar una
diferencia en las medias de los
parámetros evaluados del ± 20%,
argumento totalmente inaceptable.
De hecho una diferencia entre las
medias de los parámetros evaluados
de esa magnitud, nunca cumpliría los
criterios de bioequivalencia basados
en intervalos de confianza, como
establecen las agencias regulatorias,
incluso serían inadmisibles diferencias
entre las medias bastante inferiores a
dicho valor.
En nuestra opinión, muchos
profesionales basan su visión
negativa de los EFG en esta
consideración, que no es más que
una errónea interpretación de lo que
es un ensayo de bioequivalencia. Si
realmente fuera así, y asumiéramos
una desviación del ± 20% entre los
valores medios de las formulaciones
test y referencia, ello supondría que
por ejemplo para un valor medio
del ABC para el medicamento de
referencia (ABCreferencia) de 100
mg/L·h, la formulación que pretende
ser autorizada como EFG podría
presentar valores medios del ABC,
entre 80 y 120 mg/L·h para cumplir el
Medicamento
de referencia
0,8
1
1,25
Potenciales genéricos
No bioequivalente (bajo)
No bioequivalente (alto)
Bioequivalente
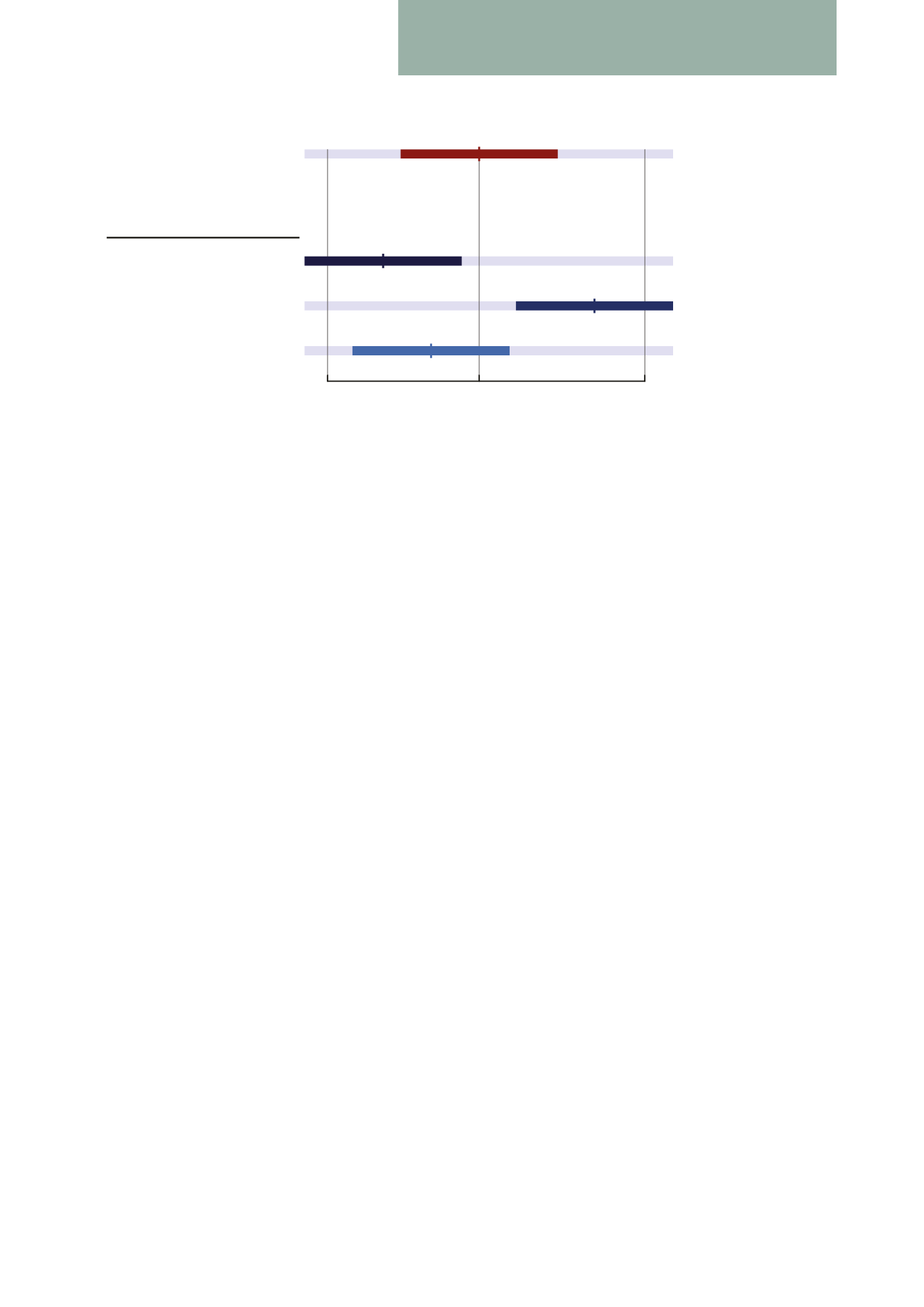
FIGURA 1.
VALORES DE IC90 DEL ABC DEL POTENCIAL GENÉRICO NORMALIZADO CON EL VALOR MEDIO DEL
ABC DEL MEDICAMENTO INNOVADOR REPRESENTADOS JUNTO CON EL INTERVALO DE REFERENCIA EXIGIDO
DE 0,80 Y 1,25 (DATOS LOG TRANSFORMADOS).
EL PAPEL DE LOS GENÉRICOS DESPUÉS DE 20 AÑOS EN ESPAÑA
BIOEQUIVALENCIA:
EL MITO DEL 20%.